
CONTENIDO
CABE ACLARAR QUE ESTE BLOG ES BASTANTE EXTENSO
PERO PUEDES ENCONTRAR FACILMENTE EL PUNTO ESPECIFICO QUE BUSCAS:
- QUE ES UN CROMOSOMA
- QUE ES UN CROMOSOMA
- ESTRUCTURA
- ENFERMEDADES DE
TRANSMISION GENETICA
- ANOMALIAS ESTRUCTURALES
CROMOSOMICAS
- CROMOSOMA 8
-
ENFERMEDADES DEBIDAS A MUTACIONES EN EL CROMOSOMA 8:
- MICROCEFALIA
-
CARCINOMA
PAPILAR DE TIROIDES
- ESCORBUTO
-
SINDROME DE
WERNER – PROGERIA
-
ACROMATOPSIA
-
LINFOMA NO
HODKIN
-
CARCINOMA
RENAL
-
LINFOMA DE
BURKITT
-
CROMOSOMA 14:
-
RETINOSIS
PIGMENTARIA
- BOCIO FAMILIAR
-
LENCEUFALOPATIA MULTIFOCAL PROGRESIVA
-
ENFERMEDAD
DE NIEMANN- PICK
-
MICROFTALMIA
-
MELOMA
CUTANEO
-
CANCER DE
PULMON
PARA RECORDAR.
Que es un Cromosoma?
Los
cromosomas (término que significa "cuerpos coloreados", por la
intensidad con la que fijan determinados colorantes al ser teñidos para poder observarlos
al microscopio), son un componente del núcleo celular que sólo aparecen cuando
la célula está en división, ya sea mitosis o meiosis; Y están compuestos por
ácidos nucleícos y proteínas.

Los
cromosomas en realidad están formados por dos cadenas
de ADN repetidas que se espiralizan
y se mantienen unidas, de forma que en un cromosoma se distinguen dos partes
que son idénticas y reciben el nombre de CROMÁTIDAS, que se unen por un punto
llamado CENTRÓMERO. El centrómero divide a las cromátidas en dos partes que se
denominan BRAZOS
Un
cromosoma es la estructura que resulta del empaquetamiento del ADN y las
proteínas previo a la división celular para su segregación posterior en las
células hijas.
El ser humano tiene 23 pares de cromosomas


El ácido nucleico (ADN), que
posee el cromosoma se divide en pequeñas unidades llamadas genes. Éstos
determinan las características hereditarias de la célula u organismo. Las
células de los individuos de una especie determinada suelen tener un número
fijo de cromosomas, que en las plantas y animales superiores se presentan por
pares.

En la célula
los cromosomas vienen en pares. Un miembro de cada par proviene de la célula
del esperma del padre y el otro miembro del par, proviene de la célula del
huevo de la madre.
En otras
palabras, el bebé recibe mitad de material genético de la madre y la otra mitad
del padre.

FORMACION DE UN CROMOSOMA DUPLICADO CON DOS CROMATIDAS
Fallas o
anomalías en la secuencia genética son causa de varios trastornos o
enfermedades de trasmisión genética,
Los errores que rara vez se producen durante la replica del DNA son los
que causan las mutaciones que sufren los cromosomas. Estas mutaciones también
pueden ser causadas por temperaturas altas, la radiación y varios compuestos
químicos. La mayoría de las anormalidades cromosómicas perjudican el organismo
que las porta. Estas mutaciones son transmitidas hereditariamente y por esta
razón, el número de personas portadoras de genes mutados
tiende a
aumentar debido a la reproducción de especies, pero también tiende a disminuir
debido a que los individuos con mutaciones genéticas no sobreviven o se
reproducen menos que sus semejantes.

Los genes
que causan mutaciones cromosómicas en su mayoría son genes dominantes. Una
persona que tenga justo una copia del gen recesivo se determina como portador,
ya que esta persona tiene el gen pero no es afectado por el. En la figura de
arriba el verde representa el gen dominante y el azul al portador.
Algunos tipos
de mutaciones cromosómicas son: la monosomía, la disomía, la trisomía, la
euploidía, la aneuploidía, la aberración cromosomica, el síndrome de down, el
síndrome de klinnefelter, el síndrome de turner, la deficiencia cromosómica, la
translocación y la inversión. Las anormalidades referidas tienden a producir
incapacidades graves dependiendo que pareja de cromosomas afecta. Como un solo
grupo, estos desordenes cromosómicos afectan a 7 de cada 1000 infantes.
Anomalías estructurales cromosómicas

Principales alteraciones estructurales de los
cromosomas.

CROMOSOMA 8
Se han descrito más de 40 casos de
duplicación invertida del brazo corto del cromosoma 8. Como en la mayoría de
los síndromes cromosómicos, el fenotipo de los niños descritos con esta
anomalía es variable e incluye anomalías faciales, agenesia de cuerpo calloso,
hipotonía, problemas neonatales de alimentación, anomalías renales y cardíacas,
múltiples anomalías menores y retraso mental grave, que es una alteración
presente en el 100 % de los casos. Muchos de los pacientes portadores de esta
cromosomopatía poseen anomalías compatibles con la vida, por lo que llegan a
adultos, aunque presentaban un importante retraso mental.
Se presentan 2 casos con duplicación
invertida del brazo corto del cromosoma 8, en los que al nacimiento se describía
una agenesia de cuerpo calloso, junto con otras malformaciones.
CASO 1
Para la comprencion del siguiente
casos se han te tener claros
algunos conceptos científicos que han sido subrayados en el texto.
COSANGUINEOS: Se aplica a la persona que desciende de los mismos antepasados que otra.
COSANGUINEOS: Se aplica a la persona que desciende de los mismos antepasados que otra.
VENTRICULOS LATERALES CEREBRALES.
Los ventrículos laterales se encuentran en los hemisferios cerebrales.
Cada ventrículo laterales compone de un cuerpo central triangular y cuatro
cuernos. Los ventrículos laterales comunican con el tercer ventrículo a través
delo que se llama el agujero interventricular

CASO 1
Recién nacido varón, producto del
tercer embarazo de unos padres sanos y no consanguíneos, ambos de 32 años. El
primer embarazo terminó en un aborto espontáneo y el segundo en una niña sana.
La gestación del caso índice fue controlada, detectándose por ecografía una
dilatación de ventrículos laterales cerebrales y una posible falta del
cuerpo calloso cerebral, por lo que a las 31 semanas de gestación, se realizó
una extracción de sangre del feto atreves del abdomen de la madre. que reveló
un "incremento de material en los brazos cortos de un cromosoma 8".
El parto fue espontáneo, a las 37 semanas de gestación, y nació vulgarmente ‘’
de cola’’.

En
el momento del nacimiento, el niño pesó 2.335 g tenía una longitud de 49 cm y
un perímetro cefálico de 31,5 . En la exploración al nacimiento se observó un
fenotipo




CROMOSOMA 8
Contiene unos 1400 genes, con 140 millones
de pares de bases: algunas de las enfermedades asociadas al cromosoma 8 son:
locus
|
Enfermedad
|
8pter-p22
|
Lipofuscinosis ceroide
|
8p23
|
Microcefalia
|
8p23.1
|
Histiocitoma
fibroso maligno
|
8p22. 16q23.3-q24.1, 13q12.11, 9q32, 3p22-p21.3, 3p22
|
Carcinoma esofágico de células escamosas
|
8p22
|
Susceptibilidad al cáncer de próstata
|
8p22-p21.3
|
Carcinoma papilar de tiroides
|
8p22-p21.3
|
Lipogranulomatosis de Farber
|
8p21.1
|
Escorbuto
|
8p12-p11.2
|
Síndrome de Werner
|
8p12
|
Síndrome de
Wolf-Hirschhorn
|
8p22-p11
|
Susceptibilidad a la esquizofrenia
|
8p11.2
|
Hiperplasia lipoide adrenal
|
8q21-q22
|
Acromatopsia
|
8q21.3-q22
|
Linfoma no Hodgkin
|
8q24
|
Anemia hemolítica debida a deficiencia de glutaton-reductasa
|
8q24.1
|
Carcinoma renal
|
8q24.11-q24.13
|
Condrosarcoma
|
8q24.12
|
Síndrome tricorinofalángico
|
8q24.3
|
Neuropatía hereditaria, tipo Lom
|
8q24.3
|
Síndrome de Rothmund-Thomson
|
8q24
|
Linfoma de Burkitt
|
8q24
|
Enfermedad de
Paget juvenil
|
Locus. Es el lugar que ocupa cada gen a lo largo de un cromosoma
(el plural es loci).

MICROCEFALIA

La microcefalia es un trastorno congénito en el cual la cabeza del bebé es
mucho más pequeña en comparación con la de un bebé normal de la misma edad y
sexo. "Micro" significa pequeño y "cefalia" se refiere a la
cabeza. La mayoría de los niños con este trastorno también tienen un encéfalo
pequeño y padecen retardo mental. Sin embargo, se debe tener en cuenta que
algunos niños con cabezas pequeñas tienen inteligencia normal.
¿Cuáles son las causas de la microcefalia?
*La microcefalia puede ser provocada por la exposición a sustancias nocivas durante el desarrollo fetal o quizás puede estar asociada con problemas o síndromes genéticos hereditarios.
*Las teorías sugieren que los siguientes factores pueden predisponer al feto a padecer los problemas que afectan el desarrollo normal de la cabeza durante el embarazo:
¿Cuáles son los síntomas de la microcefalia?
A continuación se enumeran los síntomas más comunes de la microcefalia. Sin embargo, cada niño puede experimentarlos de una forma diferente. Los síntomas pueden incluir:
*apariencia de la cabeza del bebé muy pequeña
*llanto agudo
*mala alimentación
*convulsiones
*mayor movimiento en los brazos o piernas (espasticidad)
*retardo del desarrollo
*retardo mental
*Los síntomas de la microcefalia pueden parecerse a los de otros trastornos o problemas médicos. Siempre consulte al médico de su hijo para obtener un diagnóstico.
*La microcefalia puede ser provocada por la exposición a sustancias nocivas durante el desarrollo fetal o quizás puede estar asociada con problemas o síndromes genéticos hereditarios.
*Las teorías sugieren que los siguientes factores pueden predisponer al feto a padecer los problemas que afectan el desarrollo normal de la cabeza durante el embarazo:
¿Cuáles son los síntomas de la microcefalia?
A continuación se enumeran los síntomas más comunes de la microcefalia. Sin embargo, cada niño puede experimentarlos de una forma diferente. Los síntomas pueden incluir:
*apariencia de la cabeza del bebé muy pequeña
*llanto agudo
*mala alimentación
*convulsiones
*mayor movimiento en los brazos o piernas (espasticidad)
*retardo del desarrollo
*retardo mental
*Los síntomas de la microcefalia pueden parecerse a los de otros trastornos o problemas médicos. Siempre consulte al médico de su hijo para obtener un diagnóstico.
CARCINOMA PAPILAR
DE TIROIDES

El cáncer de tiroides o carcinoma
papilar es una enfermedad que se origina en la glándula tiroides,
localizada debajo del cartílago tiroideo, en la parte delantera del
cuello. Esta glándula, en forma de mariposa, tiene dos lóbulos, el lóbulo
derecho y el lóbulo izquierdo, que están unidos por un istmo angosto.
Existen 3 tipos de tratamiento para el cáncer de
tiroides: la cirugía, el uso de yodo radiactivo y otros medicamentos
complementarios
La cirugía se realiza para extirpar la mayor cantidad de cáncer posible y cuanto más grande sea el tumor, mayor será la cantidad de glándula tiroidea que se deba extirpar. Con frecuencia, se extirpa toda la glándula.
Después de la cirugía o el yodo radiactivo, el paciente necesitará tomar un medicamento llamado levotiroxina sódica por el resto de su vida, el cual reemplaza la hormona que la tiroides normalmente produciría.
ESCORBUTO
La cirugía se realiza para extirpar la mayor cantidad de cáncer posible y cuanto más grande sea el tumor, mayor será la cantidad de glándula tiroidea que se deba extirpar. Con frecuencia, se extirpa toda la glándula.
Después de la cirugía o el yodo radiactivo, el paciente necesitará tomar un medicamento llamado levotiroxina sódica por el resto de su vida, el cual reemplaza la hormona que la tiroides normalmente produciría.
ESCORBUTO

CARENCIA DE VITAMINA C
El escorbuto es una enfermedad carencial
que resulta del consumo insuficiente de vitamina C, que es
necesaria para la síntesis correcta de colágeno en los seres humanos. El escorbuto conduce a la formación de puntos
de color púrpura en la piel, encías esponjosas y sangrado de todas las
membranas mucosas. persona afectada se pone pálida, se siente deprimida y queda
parcialmente inmovilizada. En el escorbuto avanzado se producen heridas
abiertas upurantes y pérdida de dientes. Además, empeora considerablemente
todas las demás patologías, hasta las más benignas, haciéndolas a veces
mortales.
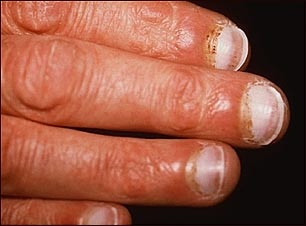
SINDROME DE WERNER – PROGERIA

.jpg){kind=link}
{kind=link}
El síndrome de werner es un desorden genético en los recién nacidos, este síndrome se
caracteriza por un rápido enviciamiento del infante, se sabe que esta
enfermedad afecta a uno de cada 8millones de recién nacidos.
La forma más severa de esta enfermedad es la llamada síndrome de Hutchinson-Gilford nombrada
así en honor de Jonathan Hutchinson, quién fue el primero
en describirla en 1886
y de Hastings Gilford quien realizó diferentes
estudios acerca de su desarrollo y características en 1904.
SÍNTOMAS
El síndrome de werner tiene varios síntomas tanto físicos como mentales:
§ Baja estatura
§ Piel
seca y arrugada
§ Calvicie prematura
§ Canas
en la infancia
§ Ojos prominentes
§ Cráneo
de gran tamaño
- sobresalientes
- Ausencia
de cejas y pestañas
- Nariz grande y con forma de pico
- Mentón retraído
-
- Problemas cardíacos
- Pecho angosto, con costillas marcadas
- Extremidades finas y esqueléticas
- Estrechamiento
de las arterias coronarias
- Articulaciones grandes y rígidas
- Manchas
en la piel semejantes a las de la vejez por mal metabolismo de la melanina
- Presencia
de enfermedades
degenerativas como
la artritis, propias de la vejez
- Muerte
natural hacia los 13 años.
- Problemas cardíacos
ACROMATOPSIA

La acromatopsia o visión sin color ocurre como
consecuencia de herencia genética y también cuando una zona de la corteza del
visual en el cerebro se lesiona
por un traumatismo cerebral. Concretamente una zona de la corteza
extraestridada, los pacientes describen su visión como similar a una película
en blanco y negro.
La acromatopsia es una enfermedad hereditaria recesiva.
La acromatopsia no es degenerativa ni conduce a la ceguera. Implica al
cromosoma X, el gen causante de la enfermedad ha sido localizado en Xq28.
Es una enfermedad poco frecuente, afecta a 1/30.000. Se puede ver pero
se pierde la capacidad de ver en color. Se utiliza también el mismo término si
se produce la visión en blanco y negro a consecuencia de un traumatismo
craneal que afecta a la corteza extraestriada.
- Acromatopsia
completa: se pierde la capacidad de ver en colores, los colores se perciben en
gamas de grises más o
menos oscuros.
- Acromatopsia
incompleta: se distingue algún matiz de color y posee
mayor agudeza visual.
LINFOMA NO HODKIN

Existen muchos tipos diferentes de linfomas no Hodgkin y se clasifican de acuerdo con la rapidez con que se propaga el cáncer.
- El cáncer puede ser de
grado bajo (crecimiento lento), grado intermedio o grado alto (crecimiento
rápido). El tumor de Burkitt es un ejemplo de linfoma de grado alto. El
linfoma folicular es un linfoma de grado bajo.
- El cáncer se subclasifica
además por la forma como lucen las células bajo el microscopio; por
ejemplo, si hay ciertas proteínas o marcadores genéticos presentes
- Sudores fríos (empapar los
tendidos de cama y el pijama aunque la temperatura ambiente no esté
demasiado alta)
- Fiebre y escalofríos intermitentes
- Picazón
- Inflamación de los ganglios linfáticos en el
cuello, las axilas, la ingle u otras áreas
- Pérdida de peso
Si el cáncer afecta las células en el cerebro, la persona puede presentar dolor de cabeza, problemas de concentración, cambios de personalidad o crisis epilépticas.
CARCINOMA RENAL

- Tratamiento con diálisis.
- Antecedentes familiares de
la enfermedad.
- Hipertensión arterial.
- Riñón en herradura.
- Poliquistosis renal.
- Tabaquismo.
- Enfermedad de von
Hippel-Lindau (una enfermedad hereditaria que afecta los vasos del
cerebro, los ojos y otras partes del cuerpo).
Síntomas
- Hinchazón y dolor abdominal
- Dolor de espalda
- Sangre en la orina
- Hinchazón de las venas
alrededor de un testículo (varicocele)
- Dolor de costado
- Pérdida de peso
- Crecimiento de vello excesivo en las mujeres
- Piel pálida
- Problemas de visión
LINFOMA DE BURKITT

- Ganglios que crecen juntos
para formar un tumor
- Ganglios linfáticos
insensibles
- Rápido crecimiento de
ganglios linfáticos
- Inexplicable inflamación de ganglios linfáticos
CROMOSOMA 14

locus
|
Enfermedad
|
14q11-q12
|
Apoptosis celular, temperatura dependiente
|
14q11.1-q11.2
|
Retinitis pigmentaria
|
14q11-q21
|
Paraplejia espástica
|
14q13.1
|
|
14q13
|
Bocio familiar
|
14q21-q22
|
Enfermedad de Hers
|
14q24
|
Leucoencefalopatía
|
14q24.3
|
|
14q24.3
|
Microftalmia, cataratas y displasia del iris
|
14q24.3
|
Tirosinemia, tipo Ib
|
14q31
|
|
14q
|
Carcinoma papilar del tiroides
|
14q32.1
|
Enfermedad oclusiva cerebrovascular
|
14q32.1
|
Leucemia/linfoma de células T
|
14q32
|
Anemia megalobástica tipo noruego
|
14q32.3
|
Melanoma cutáneo
|
14q32.33
|
Agammaglobulinemia
|
DESCRIPCION DE LAS ENFERMEDADES.
RETINOSIS PIGMENTARIA
La retinitis
pigmentaria (RP) es un grupo de desórdenes genéticos que afectan la capacidad
de la retina para responder a la luz. Esta es una enfermedad hereditaria que
causa una pérdida lenta de la visión, comenzando por una visión nocturna
disminuida y pérdida de la visión periférica (lateral). Con el tiempo, se
produce una ceguera. Desafortunadamente, no existe una cura.

La retina es la capa de células sensibles a
la luz que recubre la parte posterior del ojo, y que convierte los rayos de luz
en impulsos eléctricos. Estos impulsos son enviados al cerebro a través del
nervio óptico, donde son reconocidos como imágenes que vemos.
CAUSAS
La retinitis pigmentaria es
frecuentemente hereditaria (tiene un historial familiar). Si usted o su pareja tiene una retinitis pigmentaria,
puede haber hasta un 50 por ciento de
probabilidades de que sea transmitida a sus hijos. Si usted planea tener hijos,
pregunte a su oftalmólogo (Doctor de los Ojos) sobre una asesoría genética
Paraplejía Espástica Hereditaria
La paraplejía
espástica hereditaria es un grupo de enfermedades degenerativas genéticas de la
médula espinal, caracterizado por paraplejía o paraparesia, y rigidez o
espasticidad, con un excesivo tono muscular o hipertonía, o con sobreactividad
muscular de los músculos de las piernas

En ciertas, escasas,
familias pueden aparecer algunos de los siguientes hallazgos:
- Retraso
mental
- Demencia
- Epilepsia
- Neuropatía
periférica
- Retinopatía
- Neuropatía
del nervio óptico
- Sordera
- Ataxia
o incoordinación de los movimientos voluntarios
- Disartria
o habla dificultosa
- Nistagmo
o movimientos oculares rítmicos, involuntarios, rápidos
- Alteraciones
del sistema extrapiramidal: cambios en el tono muscular, alteración de la
postura, deterioro de la ejecución de las acciones voluntarias, movimientos
voluntarios anormales
- Ictiosis
o sequedad, engrosamiento y descamación anormales de la piel
Tratamiento
El tratamiento de la paraplejía espástica hereditaria consiste en el control médico de síntomas y medidas y equipamiento de apoyo, así como la fisioterapia. Actualmente no existe ningún tratamiento capaz de enlentecer o modificar la evolución de la enfermedad, pero la terapia con baclofén, un medicamento relajante de la musculatura esquelética puede ayudar a reducir la espasticidad en algunos pacientes. Algunos pacientes pueden ser candidatos a la quimiodenervación.
El tratamiento de la paraplejía espástica hereditaria consiste en el control médico de síntomas y medidas y equipamiento de apoyo, así como la fisioterapia. Actualmente no existe ningún tratamiento capaz de enlentecer o modificar la evolución de la enfermedad, pero la terapia con baclofén, un medicamento relajante de la musculatura esquelética puede ayudar a reducir la espasticidad en algunos pacientes. Algunos pacientes pueden ser candidatos a la quimiodenervación.
BOCIO FAMILIAR

¿Qué es el bocio?
El bocio es la
hiperactividad funcional de la glándula tiroides con hiperplasia (crecimiento),
llenándose la glándula de coloide pobre en Yodo. Si se corrige la deficiencia,
puede remitir el crecimiento. En casos crónicos persiste el crecimiento y éste
puede ser nodular.
¿Qué
la provoca?
En muchas partes
de mundo el bocio simple se debe a la carencia de Yodo y se presenta en áreas
endémicas alejadas de las costas marinas.
SÍNTOMAS
La glándula está
notablemente crecida y es palpable. Es probable que no existan síntomas o se
deban a compresión de las estructuras del cuello o el tórax: Silibancias,
disfagia, deterioro respiratorio.
Puede haber
sordera congénita recurrente (síndrome de Pendred) y trastornos del gusto.
También es posible que el paciente presente muchos síntomas de tipo nervioso.
También es posible que el paciente presente muchos síntomas de tipo nervioso.
TRATAMIENTOS
Y RECOMENDACIONES
Es más fácil
prevenir el bocio que curarlo y es más raro desde la introducción de sales
yodadas. Con la ingesta dietética de 100 a 200 microgramos de Yodo diario no
debe de presentarse el bocio simple por deficiencia.
Leucoencefalopatía multifocal progresiva

Es un trastorno
raro que ocasiona daño al material (mielina) que cubre y protege los nervios en la sustancia blanca del cerebro.
El virus JC (VJC)
causa la leucoencefalopatía multifocal progresiva (LEMP). Hacia la edad de 10
años, la mayoría de las personas han estado infectadas, pero casi nunca provoca
síntomas.
Sin embargo,
cualquier persona con un sistema inmunitario debilitado está en mayor riesgo de
desarrollar LEMP. Las causas para que se presente un sistema inmunitario
debilitado abarcan:
- SIDA (menos común ahora debido a los mejores
tratamientos para esta enfermedad)
- Ciertos
medicamentos empleados para tratar la esclerosis múltiple, la artritis
reumatoidea y enfermedades conexas
- Leucemia y linfoma
Síntomas
- Dolores de cabeza
- Pérdida de la
coordinación, torpeza
- Pérdida de la
habilidad del lenguaje (afasia)
- Pérdida de la
memoria
- Problemas de
visión
- Debilidad en
piernas y brazos que empeora
Tratamiento
En personas con
SIDA, el tratamiento para fortalecer el sistema inmunitario puede llevar a la
recuperación de los síntomas de la leucoencefalopatía multifocal progresiva. No
se ha demostrado la efectividad de otros tratamientos para esta enfermedad
Enfermedad
de Niemann-Pick

Enfermedad
de Niemann-Pick

Síntomas
- Hinchazón abdominal (área
ventral) dentro de los 3 a 6 meses
- Mancha rojo fresa en el
ojo
- Dificultades de
alimentación
- Pérdida de las habilidades
motrices tempranas (empeora con el tiempo)
- Dificultad para mover las
extremidades (distonía)
- Esplenomegalia
- Hepatomegalia
- Ictericia al nacer (o poco
después)
- Dificultades de
aprendizaje y declive intelectual (demencia)
- Convulsiones
- Mala pronunciación, habla
irregular
- Pérdida súbita del tono
muscular que puede lleva a caídas (cataplejía)
- Temblores
- Dificultad con los
movimientos oculares hacia arriba y hacia abajo (parálisis visual
supranuclear vertical)
- Marcha inestable, torpeza,
problemas al caminar (ataxia)
MICROFTALMIA

Anomalia congénita en donde se encuentra una notoria reducción del diámetro del globo
ocular en uno o en ambos ojos, generalmente hundido en una orbita tambien
pequeña.
TRATAMIENTO
Microftalmo leve asintomático: Rx
Microftalamo con Amaurosis: Protesis ocular
Criptoftalmo: enucleación o adaptación de prótesis ocular.
MELOMA CUTANEO

Se trata de
un tumor generalmente cutáneo, pero también del intestino y el ojo
(melanoma uveal) y altamente invasivo por su
capacidad de generar metástasis. A pesar de
varios años de investigaciones extensivas, el único tratamiento efectivo es la
resección quirúrgica del tumor primario antes de que
logre un grosor mayor de 1 mm.
Por lo
general, el riesgo de un individuo de contraer un melanoma depende de dos
grupos de factores: intrínsecos y ambientales. Los factores intrínsecos
incluyen la historia familiar y el genotipo heredado, mientras que el factor
ambiental más relevante es la exposición a la luz solar.[7
Prevención
y protección
Para prevenir el
melanoma, ante la llegada del verano, es preciso adoptar una serie de medidas
de protección, como la utilización de gorras o sombreros, de cremas de alta
protección, así como tomar el sol de una forma gradual y evitarlo en las horas
de irradiación más intensa (entre las 12:00 y 16:00). Incluso debajo de las
sombrillas el sol es dañino, ya que el efecto espejo de la arena puede inducir
los rayos solares con mayor intensidad
El prototipo
humano con mayores posibilidades de contraer dicha patología es una mujer entre
40 y 45 años, de piel y ojos claros que realice exposiciones solares intensas e
intermitentes desde la infancia, con quemaduras en la etapa infantil, con un
número importante de nevus congénitos o atípicos, y con antecedentes
familiares de melanoma.
Algunos consejos
para prevenir la aparición de melanomas son:
- Tomar el sol con
protección adecuada.
- Utilizar el protector solar adecuado y por todo el
cuerpo.
- Utilizar la dosis
adecuada por el fabricante.
- Debe tenerse en
cuenta a la hora de la elección del filtro tanto los rayos UVA como los UVB.
- Recurrir si fuera
necesario a la fotoprotección oral que palía las carencias y defectos de
la protección tópica.
Detección
y tratamiento
La incidencia del
melanoma en Europa, como en el resto del mundo, se ha triplicado en las últimas
décadas. Esta enfermedad representa el 75% de todas las muertes por cáncer de piel.
Para saber cuándo la apariencia es sospechosa existe una regla denominada A, B, C y D. Así, cuando un nevus es Asimétrico, tiene unos Bordes irregulares, toma una Coloración muy oscura o irregular y su Diámetro aumenta, son indicios de melanoma, por lo que se debe acudir al médico.
Se debe prestar
especial atención, por ser marcadores de melanoma, a los nevus
pigmentocelulares adquiridos que, a lo largo de la vida, modifican su
morfología. También a los nevus atípicos o(urielosis)y congénitos, siendo los
principales signos de alarma los nevus asimétricos, con bordes imprecisos,
color cambiante y sangrado. Este cuadro puede darse conjuntamente.
CANCER DE PULMON

Cromosoma 3
El cáncer de pulmón es uno de los
cánceres más comunes en el mundo. Es la principal causa de muerte por cáncer
entre los hombres y las mujeres en los Estados Unidos. El fumar cigarrillos causa la mayoría de los
cánceres de pulmón. A mayor cantidad de cigarrillos diarios que fume al día y
cuanto más joven se comienza a fumar, mayor será el riesgo de desarrollar un
cáncer de pulmón. La exposición a altos niveles de contaminación, radiación y
asbesto también puede aumentar el riesgo.
Los síntomas comunes del cáncer de
pulmón incluyen:
- Una tos
que no desaparece y empeora con el tiempo
- Dolor
constante en el pecho
- Tos con
expectoración con sangre
- Falta de
aire, silbidos al respirar o ronquera
- Problemas
repetidos por neumonía o bronquitis
- Inflamación
del cuello y la cara
- Pérdida
del apetito o pérdida de peso
- Fatiga
Existen muchos tipos de cáncer de
pulmón. Cada uno de ellos crece y se disemina de un modo distinto y se trata de
una forma diferente. El tratamiento también depende del estadio o de qué tan
avanzado se encuentre. El tratamiento puede incluir quimioterapia, radiación y
cirugía.
Casino Review 2021 - Real Players' Guide
ResponderEliminarComprehensive Casino Review 2021 먹튀 커뮤니티 – Casino 한게임 포커 Games, Bonuses, Banking Methods 포커 고수 & More. Withdrawal titanium wire speed: หารายได้เสริม 1-2 hours; Minimum deposit: €10.